- Anonymized, Secure Collection of Diagnostic Test Data and Adverse Event Tracking
- Collect Test Results Directly from “at-home” Self-Testing Patients OR from Laboratories
- Fulfil Post-Market Data Collection and Adverse Event Reporting Regulatory Requirements
- Access Up to Date,Dx Test Information Directly from the Manufacturer
- Securely & Confidentially Capture, Store and Share your Test Data
- Own your Health Data
- Monetize your Health Data while Protecting your Identity
- Test-to-Treat: For patients to quickly access state-of-the-art testing information and clinical care
- Test-to-Validate: For Dx test manufacturers to reach a large cohort of consumers, disseminate accurate testing information, and secure post-market data collection to obtain FDA clearance
- Test-to-Track: For public health agencies to monitor nation-wide disease prevalence, clinical symptoms in different patient cohorts, new outbreaks, etc.
A former FDA IVD regulatory scientist and a Leader in innovative software development met the FDA’s challenge of capturing COVID-19 diagnostic test (Dx) data from anywhere. The platform that we developed won the FDA’s Design-a-thon and was successful in capturing test data from individuals as well as organizations outside the standard healthcare system. We have now expanded our platform beyond COVID testing to help Dx test developers to disseminate their curated product information to consumers and to collect test results and relevant information directly from the consumers themselves or via an approved laboratory setting. With the roll out of the new FDA regulatory requirements for LDTs, we are well equipped to help you to work with CDRH to fulfil your obligations.
National Human Genome Research Institute
Policy and Program Analysis Branch
If implemented, the following timeline would ensue:
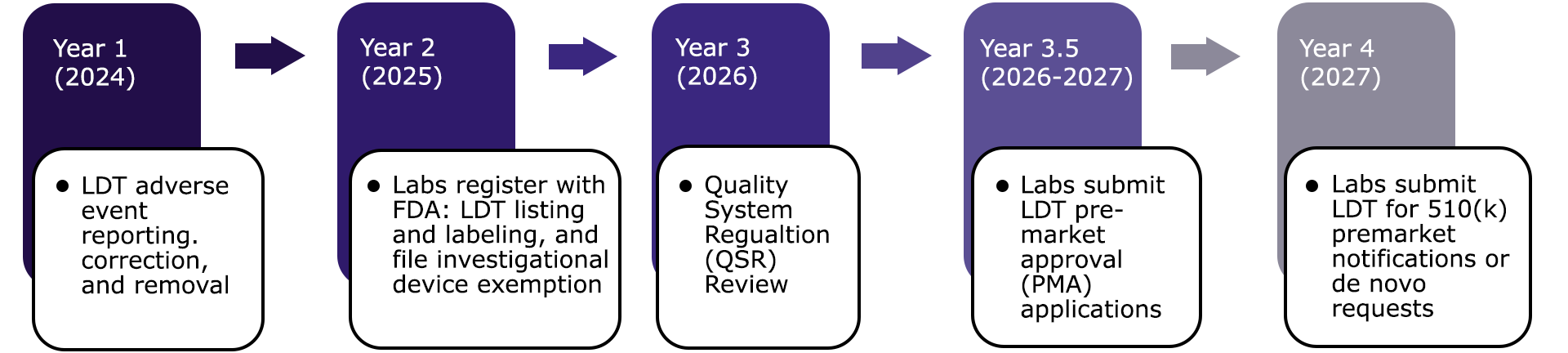
- 2024: Labs would be required to submit LDT reports including adverse events that have or could have caused harm. FDA reviews the reports and decides what tests should be corrected or removed.
- 2025: Labs conducting LDTs must register with the FDA and list all LDTs, and file investigational device exemptions (IDEs)
- 2026: LDTs and labs would undergo Quality System Regulation (QSR) Review, which is used to assess device manufacturers that intend to distribute medical devices commercially.
-
2027: Labs would submit pre-market approval applications, a process in which the FDA conducts a scientific and regulatory review of the LDT to determine safety and effectiveness.
- After pre-market approval, labs submit either 510(k) or de novo requests to the FDA for full approval. A 510(k) application is designed for tests with a predicate on the market, while de novo applications are for tests without an analog on the market.